先発医薬品とほぼ同じ効果を持つとされるジェネリック医薬品。しかし、単に成分が同じであればいいわけではありません。米国でジェネリック医薬品を販売するためには、 生物学的同等性試験(Bioequivalence Study)という非常に厳しいハードルを越え、FDA(米国食品医薬品局)にその性能を証明する必要があります。もしこの証明が不十分であれば、どれだけ高品質な薬を作ったつもりでも、市場に出ることは不可能です。
では、FDAは具体的に何をチェックし、どのようなデータがあれば「同等である」と認めるのでしょうか。ここでは、メーカーが直面する審査の基準から、コストを削減できる特例措置まで、実務的な視点で詳しく解説します。
生物学的同等性とは何を意味するのか
まず整理しておきたいのが、FDAが定義する「生物学的同等性」の中身です。簡単に言うと、 生物学的同等性は、同じ用量の薬を同じ条件下で投与したとき、有効成分が吸収される速度(Rate)と量(Extent)に、統計的に意味のある差がない状態を指します。つまり、体の中での「動き」が先発薬と同じであることを証明しなければなりません。
FDAへの承認申請である ANDA(後発医薬品承認申請)を通過するには、以下の2点を同時に満たす必要があります。
- 製剤的同等性:有効成分、剤形、含量、投与経路が先発薬(参照薬:RLD)と同一であること。
- 生物学的同等性:血中濃度などのデータで、体内動態が先発薬と同等であること。
この考え方の根底には、「生物学的に同等であれば、治療効果や安全性も同等である」という基本的な仮定があります。このため、ジェネリックメーカーは改めて大規模な治験(第III相試験など)を行う必要がなくなり、この試験さえクリアすれば承認への道が開ける仕組みになっています。
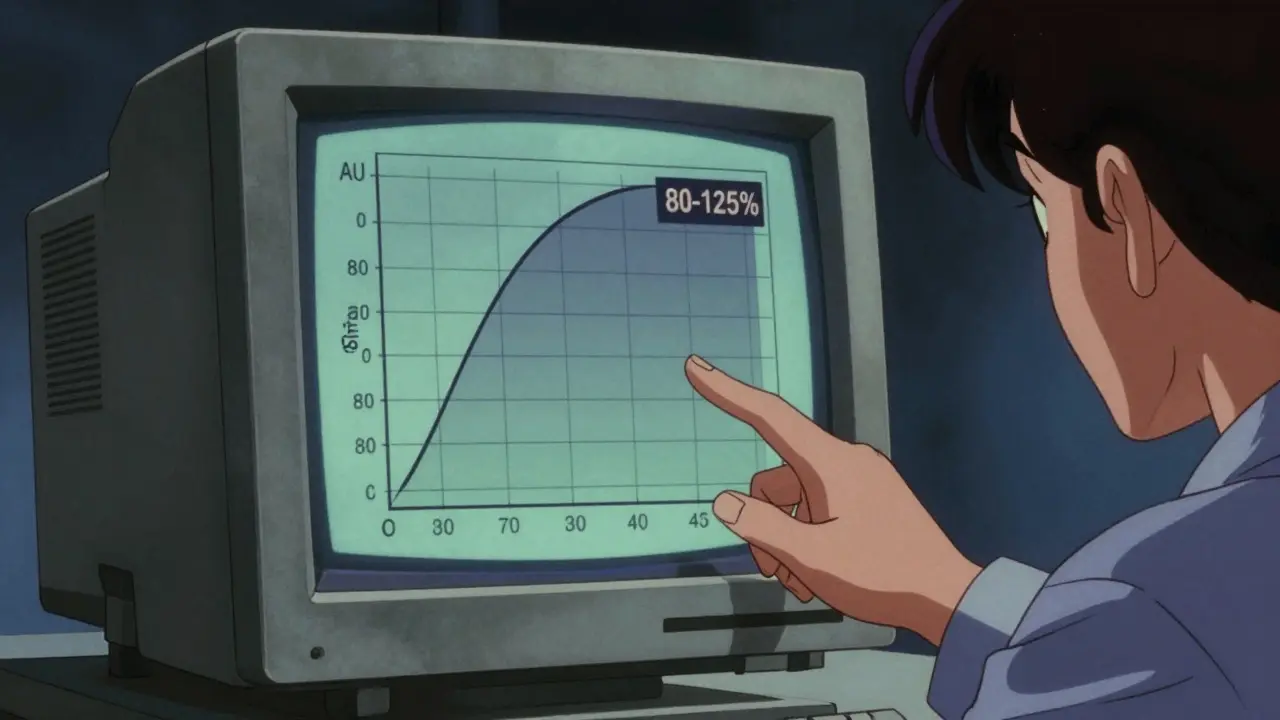
FDAが課す「80-125%ルール」の正体
FDAの審査で最も重要視されるのが、数値による客観的な証明です。具体的には、血中の薬物濃度を測定し、以下の2つの主要指標をチェックします。
- AUC(血中濃度-時間曲線下面積):成分がどれだけの量、体内に吸収されたかを示す指標。
- Cmax(最高血中濃度):成分が最大でどこまで濃度が上がったかを示す指標。
ここで登場するのが、業界の金字塔とも言える「80-125%ルール」です。ジェネリック薬(テスト薬)と先発薬(参照薬)の幾何平均値の比率を算出し、その90%信頼区間が 80% から 125% の範囲内に収まっていることが求められます。
| 評価指標 | 判定基準(90%信頼区間) | 意味合い |
|---|---|---|
| AUC | 80% ~ 125% | 吸収された総量が同等か |
| Cmax | 80% ~ 125% | 吸収されるスピードと最大濃度が同等か |
ただし、すべての薬がこの基準で良いわけではありません。例えば、わずかな濃度の差が劇的な副作用や効果不足につながる 狭い治療幅を持つ薬(NTID)(例:ワルファリンやレボチロキシン)の場合、FDAはより厳しい 90-111% という極めてタイトな基準を適用することがあります。これは、患者の安全性を最優先にするための措置です。
試験の実施方法と直面するコストの壁
生物学的同等性を証明するための試験は、通常、24〜36人の健康な成人ボランティアを対象に行われます。多くの場合、空腹状態で投与され、厳格なGLP(Good Laboratory Practice)基準に従ってサンプル管理が行われます。場合によっては、食事の影響を調べる「食後試験」も追加で要求されます。
メーカーにとって最大の悩みは、この試験に莫大なコストがかかることです。1つの試験につき、およそ50万ドルから200万ドル(数千万〜数億円)の費用が発生すると言われており、開発予算を圧迫します。また、サンプル数や分析手法に不備があると、FDAから差し戻しを受け、さらなる追加費用と時間が発生します。
実際、ANDAの初回申請での承認率は約43%に留まっており、その主因の多くがこの生物学的同等性データの不備にあります。一方で、FDAが発行している 製品別ガイダンス(PSG)に厳密に従った企業は、初回承認率が68%まで跳ね上がるというデータもあり、事前準備の重要性がわかります。
in vivo試験を回避できる「バイオウェイバー」とは
人間を対象としたin vivo(生体内)試験は時間もコストもかかります。そこで、特定の条件を満たせば試験を免除してくれる制度が バイオウェイバー(Biowaiver)です。これは、物理化学的な特性がほぼ同一であれば、わざわざ人体で試さなくても同等であると見なす仕組みです。
バイオウェイバーが適用される主な判断基準には「Q1-Q2-Q3フレームワーク」があります。
- Q1(定性的同等性):有効成分および添加剤が同一である。
- Q2(定量的同等性):剤形と濃度が同一である。
- Q3(物理化学的同等性):pHや溶解度などの特性が同等である。
例えば、静脈注射用の溶液や、局所的に作用する点眼液、点耳薬などが対象になりやすい傾向があります。また、皮膚に塗る外用剤などの場合は、in vivo試験の代わりにin vitro(試験管内)での放出試験(IVRT)や透過試験(IVPT)で代用できるケースもあります。バイオウェイバーをうまく活用できれば、承認までの期間を6〜12ヶ月も短縮できる可能性があります。
複雑なジェネリック医薬品への新しいアプローチ
最近のトレンドとして、吸入剤や経皮吸収製剤のような「複雑なジェネリック医薬品」への対応が挙げられます。これらは単純な血中濃度測定だけでは同等性を証明するのが難しいため、FDAは新しい手法を導入しています。
その一つが、 PBPKモデリング(生理学的薬物速度論モデリング)の活用です。これはコンピュータ上のシミュレーションを用いて、薬が体内でどう動くかを予測する手法で、試験の効率化や設計の最適化に役立てられています。
また、FDAはGDUFA III(ジェネリック医薬品ユーザーフィー法)のもと、複雑な製剤に対する新しいガイダンスを次々と発行しています。これにより、これまで「証明不能」として断念していた製品でも、適切な評価指標(エンドポイント)を設定することで承認を得られる道が開かれつつあります。
生物学的同等性試験はすべてのジェネリック薬で必要ですか?
いいえ、必ずしもすべての製品でin vivo試験が必要なわけではありません。先述のバイオウェイバー(試験免除)の基準を満たす製品や、特定の物理化学的試験で代用可能な製品については、人間を対象とした試験を省略できる場合があります。
「80-125%ルール」に外れたら、その薬は危険なのですか?
必ずしも「危険」ということではありませんが、「先発薬と同じ効果が得られる保証がない」ということになります。例えば、吸収が早すぎれば副作用が出やすくなり、遅すぎれば十分な治療効果が得られないリスクがあります。そのため、FDAはこの範囲を厳格に運用しています。
ANDA申請で生物学的同等性が原因で却下される主な理由は何ですか?
主な原因には、不適切な試験デザイン、サンプルサイズの不足、分析手法の精度不足、そして不完全なドキュメント作成が挙げられます。特に製品別ガイダンス(PSG)を無視して独自の設計を行った場合に、不備を指摘される確率が高まります。
CmaxとAUCのどちらがより重要視されますか?
どちらか一方が重要というわけではなく、両方を満たす必要があります。AUCは「全体の吸収量」を、Cmaxは「吸収の速さ(ピーク)」を評価するため、例えば即効性が求められる薬ではCmaxが、持続的な効果が求められる薬ではAUCが特に重要な意味を持ちますが、承認には両方のクリアが必須です。
FDAの審査期間を短縮する方法はありますか?
製品別ガイダンス(PSG)に忠実に従うことが最も確実な近道です。また、米国内で製造されたAPI(医薬品有効成分)を使用し、米国内で生物学的同等性試験を実施した製品に対して優先的な審査を行うパイロットプログラムなども検討に値します。
次のステップ:メーカーが取るべき戦略
これからFDA承認を目指すメーカーにとって、最もリスクを減らす方法は「徹底した事前調査」です。まずは自社製品がバイオウェイバーの対象になるかを確認し、対象外であれば最新の製品別ガイダンス(PSG)を熟読してください。もし複雑な製剤を扱うのであれば、PBPKモデリングなどの最新ツールを導入し、試験の失敗確率を下げるシミュレーションを行うことが推奨されます。
また、分析方法のバリデーション(妥当性確認)を疎かにせず、GLP準拠のデータ管理を徹底することで、レビューサイクル(審査の往復)を最小限に抑え、市場投入までの時間を短縮できるはずです。










tomomi nakamura
4月 7, 2026 AT 16:33バイオウェイバーの仕組みって合理的ですね。物理化学的な特性が同じならわざわざ人体試験をしなくていいっていうのは、効率面でも倫理面でも理にかなってる気がします。
YOSUKE MASU
4月 8, 2026 AT 04:29結局はFDAのガイドラインという「正解」にどれだけ忠実になれるかって話だよね。独創性なんてこの業界ではリスクでしかない。笑えない現実だわ ┐(´∀`)┌
kazumi sakurai
4月 9, 2026 AT 15:55は?80-125%で十分だって本気で言ってんの?この緩い基準で「同等」とか笑わせないでほしいんだけど。個体差があるのに統計的な平均でごまかしてるだけで、現場の患者がどうなるか全然考えてないでしょ。FDAの審査なんて結局は書類仕事のやり取りだけで、中身はスカスカなのが見え見え。こんなガバガバな基準で薬を流通させてる時点で、業界全体のレベルが低すぎるわ。
Noriyuki Kobayashi
4月 11, 2026 AT 10:28ANDA申請における初回承認率の低さは、まさに準備不足の表れと言わざるを得ません。製品別ガイダンス(PSG)を徹底的に分析し、戦略的に試験設計を行うことが、開発期間の短縮とコスト抑制の唯一の道であると確信しております。地道な努力こそが最善の策です。
ゆうや とみおか
4月 13, 2026 AT 04:08あーはいはい、PSGに忠実なら承認率が上がるなんて最高の攻略本ありますねーw 開発予算数億円飛ばして「やっぱり不備がありました」って泣きつくメーカーが目に浮かぶわ。最高に効率的なシステムだことw
Mayumi Uchida
4月 14, 2026 AT 20:08数値的な同等性が、果たして真の意味での「同一性」を担保し得るのかという点に、深い思索を促されます。科学的な近似値による証明は便宜的な合意に過ぎず、生命という複雑系における個別の反応までを完全に制御することは、人間の傲慢さという側面を内包しているのではないでしょうか。形式的な基準の充足が、必ずしも本質的な救済に結びつくとは限らないという逆説に、我々は向き合うべきかと存じます。
yuu tsuda
4月 15, 2026 AT 20:13なるほど〜、数億円かけて試験して半分近く落ちるなんて、なんて素晴らしいビジネスモデルなんでしょうねぇ✨ 企業の資金を効率よく溶かすための完璧なシステムで感動しちゃいます💖 ああ、本当に素晴らしい世界ですね😊
Ayana Women's Wellness
4月 16, 2026 AT 04:30PBPKモデリングみたいな最新ツールを使いこなせれば、失敗のリスクをガッツリ減らせますよね!今の時代、泥臭い試験だけじゃなくてデジタルでのシミュレーションを組み合わせるのが勝ち筋。みんなで最新技術を武器に、もっと安心安全な薬を早く届けられるように頑張りましょう!応援してます!🌈
Haru Chiaki
4月 18, 2026 AT 00:0490-111%っていうタイトな基準があるなら、そこだけ徹底して他は適当にやりましょうよ。どうせ多くの人は気付かないし、書類さえ整っていればFDAもOKしてくれるでしょ。効率化ってそういうことじゃないの?
Hana Hatake
4月 18, 2026 AT 16:51NTID(狭い治療幅を持つ薬)への厳しい基準適用は、安全性への配慮として適切であると考えられます。